文献学习2024-BSAlign比对算法
文献题目:A Library for Nucleotide Sequence Alignment
doi:https://doi.org/10.1093/gpbjnl/qzae025
背景:中国农业科学院深圳农业基因组研究所(岭南现代农业科学与技术广东省实验室深圳分中心)阮珏团队和邵浩靖团队开发了一种DNA比对新技术“BSAlign”,相比较同类并行算法,该算法可更快生成最优比对结果,且准确性更高。相关研究成果以题为“BSAlign: A Library for Nucleotide Sequence Alignment”发表在 基因组蛋白质组与生物信息学报(Genomics, Proteomics & Bioinformatics(GPB))
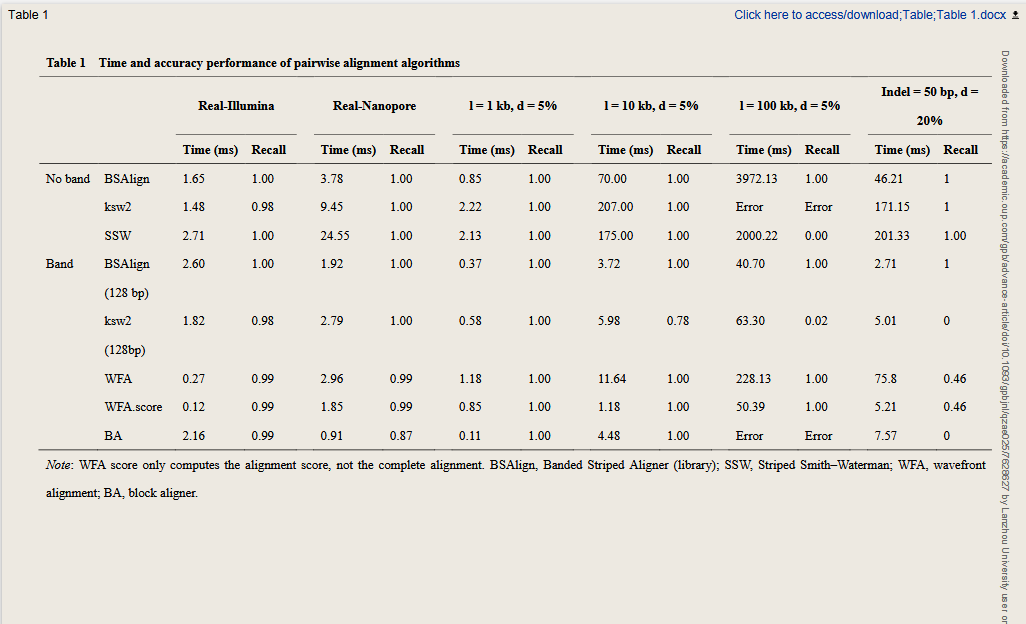
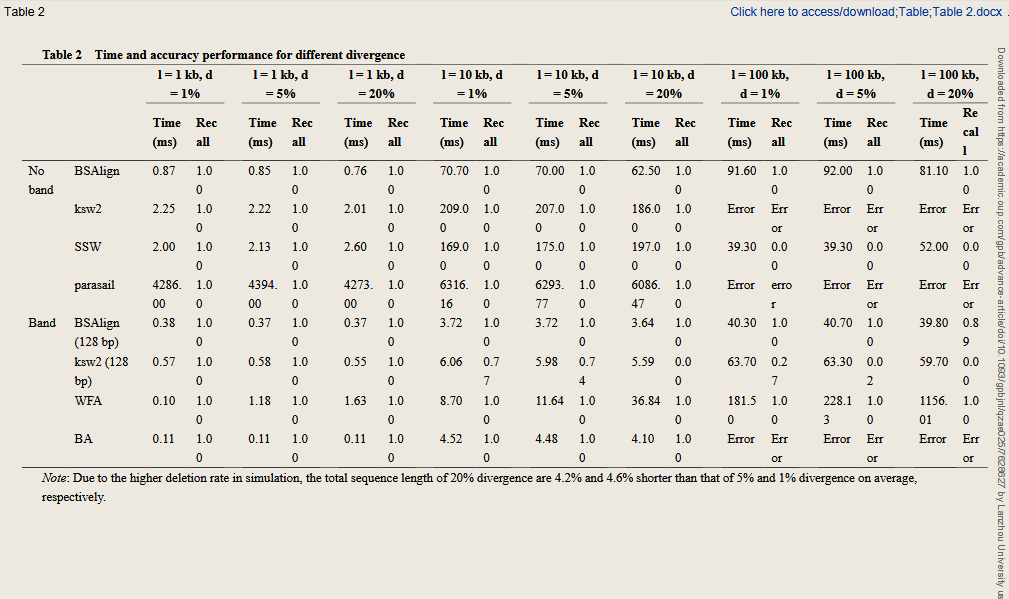
主题:经典的动态规划算法,如史密斯-沃特曼算法和尼德曼-翁施算法,常用于处理序列比对,但由于其时间复杂度呈二次函数式增长,当序列长度增加时,算法的处理时间也随之变长,导致其在处理大规模序列比对时效率低下,严重阻碍了其在大规模序列比对中的应用。目前并行加速比对的最优算法有三种方法:通过增加数据并行度获得加速的条纹法;通过减少计算单元的字节数从而增加并行度的差分法;通过减少整体计算量获得加速的带宽法。然而,目前并没有任何方法可以高效地结合这三种方法,获得更快速的比对算法。研究人员提出了条纹移动法,该算法在带宽环境下实现了高效运算,并开发了主动F循环法,解决了条纹数据在长插入或删除情况下的多次查询问题。这一创新显著提高了比对速度。与现有并行算法相比,BSAlign比对算法的速度提升了2倍,在长序列比对方面,其效率较基于编辑距离的比对算法提高了1.5到4倍。
优点:
目前加速比对方法:
- 通过增加数据并行度获得加速的条纹法;
- 通过减少计算单元的字节数从而增加并行度的差分法;
- 通过减少整体计算量获得加速的带宽法。
BSAlign创新点:
提出了条纹移动法,该算法在带宽环境下实现了高效运算
并开发了主动F循环法,解决了条纹数据在长插入或删除情况下的多次查询问题
速度有优势
经典算法:
Needleman-Wunsch算法和Smith-Waterman算法。他们通过解决 动态规划 (DP) 问题来处理序列比对,其中计算评分矩阵并返回来自具有最大分数的单元的最佳路径。虽然这两种方法在寻找最佳比对结果方面表现出了很强的能力,但它们需要二次方的时间复杂度并且快速退化,尤其是在处理长序列时。(具体参考生物信息学课本讲解,如果没记错两种算法大致是全局和局部的画箭头规划走路,再加上空位罚分之类的)
准确性:
相比其他几种算法准确性较高
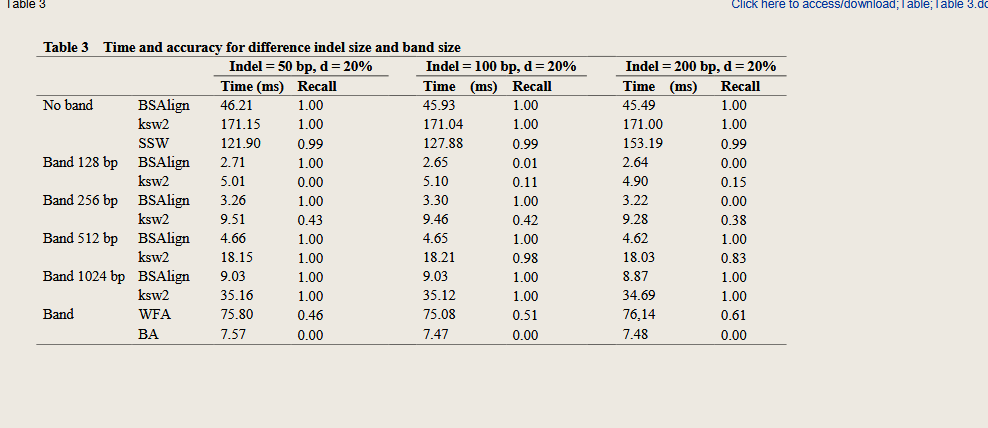
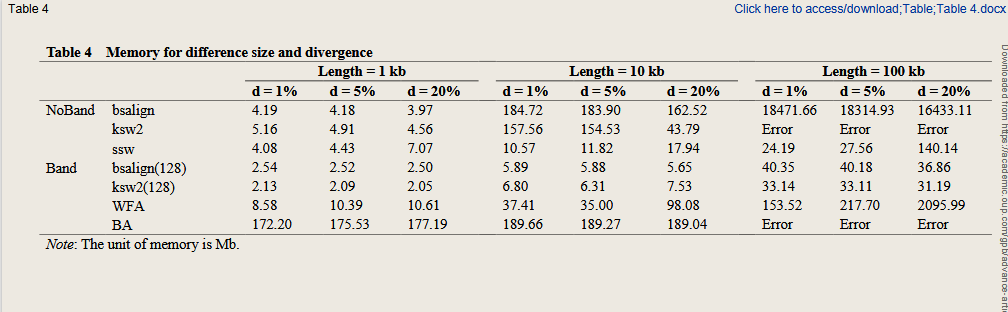
原理:
单指令多数据(SIMD): 第一个优化类别是重新设计评分矩阵计算的数据结构,解决相邻单元之间的数据依赖关系,从而消除 DP 算法内循环内的条件分支,SIMD 等并行化技术更加高效。
在该类别的最初 个试验中,Wozniak 提出了一种与次对角线平行存储值 的实现,以消除 个传统实现的内循环中的条件分支,并实现了 2 倍的加速。
在另一项试验中,Rognes 等人。 引入了另一种实现来存储与查询 序列并行的值。与 Wozniak 的实现相比,Rognes 的设计 的一个优点是它只需要为整个参考序列计算一次查询配置文件。然而,缺点是在计算 F 矩阵时,条件分支被放置在内部循环中。
对于最近的工具,例如 BGSA 、SeqAn 和 AnySeq ,单个指令的长度范围从 128 位到 512位。
F evaluation
为了结合 Wozniak 和 Rognes 的优点,Farrar 修复了这些内容,引入与 SIMD 寄存器并行但以条带模式访问的查询序列布局的缺点,该布局仅计算查询配置文件一次 并将条件 F 矩阵评估移至内部之外环形。结果,Farrar 的条纹矢量化成功地加速了 Smith–Waterman 算法 并被许多对准器采用,例如 Burrows–Wheeler Alignment Smith–Waterman (BWA-SW) 、Bowtie2 和条纹史密斯-沃特曼 (SSW) 库 。然而,同一寄存器中的单元并不总是彼此独立的。 Farrar 通过为每个 F 元素添加一个校正循环解决了这个问题,当插入/缺失足够长时,该循环可能会迭代多次。
下面是github安装使用介绍
bsalign
ruanjue/bsalign: Banded Striped DNA Sequence Alignment (github.com)
Installation
1 | git clone https://github.com/ruanjue/bsalign.git |
run bsalign
1 | bsalign |
1 | commands: |
Example
1 | cd example |
Result Example
1 | 29.1 75 + 0 75 29.2 76 + 0 76 128 0.934 71 4 0 1 |
Result Format
Each result is 4 lines. Line 1 :col1-RefName; col2-RefLength; col3-RefStrand; col4-RefStart; col5-RefEnd; col6-QueryName; col7-QueryLength; col8-QueryStrand; col9-QueryStart; col10-QueryEnd; col11-AlignmentScore; col12-Identity; col13-NumberOfMatch; col14-NumberOfMismatch; col15-NumberOfDeletion; col16-NumberOfInsertion Line 2 :Reference Sequence Line 3 :’|’, ‘*’ and ‘-‘ mean match, mismatch and indel, respectively. Line 4 :Query Sequence
use bsalign library
copy bsalign directory into your code cp -r /path/to/bsalign .
Pairwise Alignment Example
1 | #include "bsalign/bsalign.h" |
Multiple Alignment Example
1 | #include "bsalign/bspoa.h" |